
Kevin Taliaferro; Sigurd Berven
INTRODUCTION
Paget’s disease is a disorder of bone metabolism that results in an abnormal bone turnover and remodeling. The effect of Paget’s disease on bone leads to compromise in the strength of cancellous bone architecture and may cause pain, risk of fracture and deformity. Paget’s disease of bone most commonly is symptomatic in the axial skeleton, including the spine and pelvis.
Writing to the Medical and Chirurgical Society in November 1876, English surgeon Sir James Paget first described the disease that would later be named after him. He presented five cases of this novel disease from his practice, including one patient he had followed for more than 20 years. Although he would erroneously describe it as a polyostotic inflammatory condition called osteitis deformans, his defining characteristics of the disease were quite prescient. “It begins in middle age or later, is very slow in progress, may continue for many years without influence on the general health, and may give no other trouble than those that are due to the changes of shape, size, and direction of the diseased bones. Even when the skull is hugely thickened, and all its bones exceedingly altered in structure, the mind remains unaffected.”1
In addition to his excellent clinical description, his histologic description and hand drawn image of the microscopic appearance of the diseased bone are quite exquisite (see Fig. 3-1). His description reads like a modern-day pathology report. “With a medium power the number of lamellae surrounding the Haversian canals was easily seen to be not larger than in normal bone, whilst the arrangement of the intervening space was most complex and totally different from that of healthy bone.”1

Although the full nature of Paget’s disease has not been fully elucidated, we do know that Paget’s disease is a focal dysregulation of bone turnover in one or several bones that mainly affects 40 to 50-year-olds of western European descent. The purpose of this chapter is to review the pathophysiology, epidemiology, clinical presentation, the evaluation and management of Paget’s disease of bone in general, and its effect on the spine in particular.
ETIOLOGY
Environmental
Due to its regional prevalence differences, Paget’s Disease (PD) has long been thought to be secondary to environmental factors. Paget’s has been previously found to have a positive association with dog ownership.2 Initially, this claim was refuted by other studies that found no association.3 However, other studies have focused on, specifically, owners of unvaccinated dogs and have found a significant increase in the risk of developing PD if a dog is not vaccinated against canine distemper.4 Reverse-transcriptase polymerase chain reaction and in situ hybridization techniques have isolated canine distemper RNA from Pagetic osteoclasts and osteoblasts.5,6 Canine bone marrow cells infected with canine distemper virus (CDV) in vitro were observed forming multinucleated cells, similar to the phenotype seen in PD osteoclasts. In human cell lineages, preosteoclasts infected with CDV were found to increase in number, size and bone resorption.7 In addition, CDV infection induced a dramatic increase in the expression of NF-kB and SQSTM1/p62.
Chemical exposure has also been postulated as a potential cause of PD.8 A cluster of towns in the Lancashire region of England were found to have an abnormally high prevalence of PD. Accounting for time lag to disease presentation, the author found a positive association with high levels of arsenic used by the cotton industry in the region between 1917 and 1945. Subsequent prohibition of the use of arsenic in later decades was associated with a decline in PD prevalence in the area by 1993.
Viral
Dysregulated osteoclast function has long been agreed to be the primary cause of Paget’s disease. However, the etiology of this dysregulation continues to be the subject of debate. First described in 1974 by Rebel et al. and Mills et al. in 1976, osteoclasts of patients with Paget’s disease were found to have nucleocapsid inclusion bodies in the nucleus and cytoplasm, suggesting a possible paramyxoviral etiology.9,10 Interestingly though, none of these inclusion bodies have been found in osteoblasts or osteocytes, suggesting that these inclusion bodies may be a red herring rather than a true finding. Several other diseases have been reported to have similar nuclear inclusion bodies in multinuclear osteoclasts, such as familial expansile osteolysis,11 Pycnodysostosis12 and Osteopetrosis.13 Narrowing down a specific paramyxoviral source has been more elusive. Several paramyxovirus antigens have been isolated including respiratory syncytial virus, parainfluenza, canine distemper and measles.14-18 However, no intact virions have been detected budding from the cell membrane, suggesting the viruses are defective. Although no intact viruses have been seen, the fact that paramyxoviruses are negative-sense RNA suggests that there is a lack of integration into the host genome.
Rivera-Toledo et al. showed that respiratory syncytial viruses, which are paramyxoviruses, are able to evade a host’s immune system and achieve a non-lytic state in murine macrophages.19 Not only does this allow the infected cell to continue to be a viral reservoir, but the infection has been shown to alter the expression of genes coding for pro-inflammatory cytokines, for trans-membrane proteins related to antigen uptake, and for those proteins related to cell survival without integration into the host’s DNA.
Genetics
Genetics has been a focus of investigation recently, as there appears to be a familial link in approximately one third of Paget’s disease cases. Patient’s with a first-degree relative with Paget’s disease are seven to ten times more likely to develop the disease than the general population.20 When looking at familial clusters of the disease, it appears that it has an autosomal dominant pattern of inheritance with an elevated but incomplete penetrance with increasing age.21,22 Genome-wide linkage analysis of these familial clusters has found several target chromosomal loci, including 2q36, 5q31, 5q35, 10p13, and 18q21.23-25 So far, 5q35 loci has shown the strongest evidence of genetic linkage. Both Laurin et al.24 and Hocking et al.25 demonstrated mutations in the SQSTM1 gene were responsible for 5q35 linked Paget’s disease. The SQSTM1 gene codes for the p62 protein, which plays a non-fully-elucidated role in the NFkB signaling pathway–essential for osteoclast maturation, function and survival. SQSTM1 mutations have been found in approximately 40% of individuals with familial PD. In addition, individuals with this mutation of the disease have been shown to have earlier onset, a greater number of bones affected and higher severity.26-30
Several genome-wide association studies have looked at the role other loci play not only in the presentation, but in the severity of Paget’s disease. Seven loci have been identified across SQSTM1-negative populations from around the world that appeared to be associated with severity of the disease. The greater the number of these alleles these patients had the more extensive their disease. The proportion of familial risk of these combined seven loci is approximated at 13%. Albagha et al.31 weighted each allele with its effect size to produce a risk allele score. They found that those patients with the top 10% risk score were 10 times more likely to have Paget’s disease than those in the bottom 10%. However, the combined effect of these seven loci was still far lower than the effect size of the SQSTM1 mutations.31-33
The identification of these multiple genetic variations brings with it the potential for screening of individuals. With its high prevalence, penetrance and effect size, SQSTM1 would be an ideal screening target. Zoledronate in the Prevention of Paget's or the ZiPP trial, which is currently underway, is offering genetic testing for children of patients with Paget’s Disease to screen for SQSTM1 and then randomizing them into treatment groups of zoledronic acid or placebo. Ideally, testing of multiple alleles associated with the disease will allow for individuals to be given a risk score and earlier surveillance in hopes of staving off advanced disease with late presentation.34
EPIDEMIOLOGY
Paget’s disease is the second most common metabolic/bone remodeling disease, following only osteoporosis. Males are found to have a slightly higher preponderance of the disease. It is far more common in the elderly. Patients older than 85 have a five-times higher incidence than those under 65. Its prevalence is estimated to be one to three percent of European white adults over the age of 55. Archaeologic studies have even shown the disease to be present in Europe during the period of the Roman Empire.35 Furthermore, skeletal studies across multiple time periods and regions in Europe show a higher prevalence of skeletons with PD in British populations. This suggests that the disease most likely emanated from Britain with dissemination through human migration.36 This theory is reinforced by contemporary studies showing a strong link between PD prevalence in England and western Europe, as well as in New World countries that experienced a significant influx of immigrants from those places. This includes the United States, Canada, New Zealand and Australia. The disease remains relatively rare in Asia, the Middle East and Africa.37
Interestingly, epidemiology of PD shows some significant changing trends in the last century. First, longitudinal studies demonstrated a decrease in the prevalence as well as the severity of PD. Cundy found an approximately 50% reduction of the prevalence of PD in New Zealand between 1973 and 2002.38 These findings were similarly echoed in Cooper et al.’s paper that found a significantly steep decline in Britain between 1974 and 2004.39 Secondly, Cundy also found that the average age of presentation was significantly later, at 62 to 74 years of age.38 The true meaning of these findings, however, is unclear. Being cross-sectional population studies, there is a risk of bias affected by changing populations during this time. With increasing world travel, the examined populations may have been more ethnically heterogenous at later time points. However, proponents of these findings suggest that changing environmental factors may explain these trends.
CLINICAL PRESENTATION
Orthopaedic Presentation Broadly
Paget’s disease can be quite insidious in its development and is frequently found by coincidence on radiographs performed for other purposes. It is estimated that more than two-thirds of people with Paget’s disease are asymptomatic. If they have low-grade symptoms due to the age of presentation, they can be frequently given a non-specific diagnosis of age-related changes, leading to a delay in diagnosis. The disease is most commonly polyostotic (65-90%) but can be monostotic in some cases. In polyostotic patients, the disease appears to arise at the same time point and will be limited to the original bones in which it appeared. The most common sites of involvement are the pelvis, spine and skull. However, disease can be seen in any bone and frequently involves the femur, tibia and humerus.
Generally, presenting symptoms can be highly variable but most frequently include bone pain, osteoarthritis, deformity and/or fracture. Bone pain is the most common complaint. Pain is frequently is worse at nighttime or while at rest. However, in the presence of osteolytic lesions pain may be worsened with weight bearing.
Although osteoarthritis is neither unique to PD nor uncommon in the older age groups, it is a common finding with progression of the disease. Involvement of the subchondral bone as well as altered joint biomechanics from osseous expansion can lead to rapid degeneration of the articular surface. The hip and the knee joints are reported to be the most commonly involved joints.
Deformity of the long bones of the limbs was one of the first hallmark features defined by Paget. In his paper, Paget described his patient’s progressive deformity—“The left femur and tibia became larger, heavier, and somewhat more curved. Very slowly those of the right limb followed the same course, till they gained very nearly the same size and shape. The limbs thus became nearly symmetrical in their deformity, the curving of the left being only a little more outward than that of the right.”1
Deformity of the bones is now understood to be due to disorganized and unbalanced bony remodeling. Abnormal osteoclastic function is found at the endosteal surface of the bone, leading to asymmetric resorption. At the periosteal and endosteal surfaces, overactive osteoblasts lead to appositional growth. Combinations in these remodeling patterns lead to the progressive disfigurement of the bones involved. There are four types described: periosteal and endosteal apposition, periosteal apposition and endosteal absorption, periosteal apposition with unchanged endosteum, and focal periosteal apposition.
Spine-specific Pathology
The clinical presentation of Paget’s disease in the spine is unique in that it not only has the associated bone symptoms, but that neurologic symptoms can arise as well. One of the most common presenting complaints is low back pain, reported in as many as 54% of cases.40 However, this presentation can be clouded by Paget’s disease occurring in an age group that frequently has high levels of coexisting osteoarthritis. When Altman et al. looked at this question, they could attribute back pain to Paget’s disease in only 12% of cases. Additionally, one-third of patients present with signs of spinal stenosis.41
RADIOGRAPHIC FINDINGS
Plain Film
Three characteristic phases of Paget’s disease have been described using plain radiography. The initial lytic phase is characterized by a sharp advancing edge of osteolysis. This is referred to frequently as a “blade of grass” sign. In the intermediate or mixed lytic/blastic phase, there are signs of both lytic and blastic processes occurring simultaneously. The main defining characteristic that defines this phase, however, is new bone formation. Trabeculae begin to take on a more coarsened appearance. Cortex expansion and thickening occurs at this time as well. It is in this phase that the dimensions of the vertebrae also begin to change. Anterior to posterior vertebral body expansion is noted as well as enlargement of the posterior elements. There is a loss of the normal anterior convexity of the body. Since there is no periosteum/endosteum interface at the endplates, the heights of the vertebral bodies are not increased. The late, or sclerotic, phase is associated with outward signs of the disease including progressive deformity secondary to fractures and abnormal repair. Involved bones show marked osteosclerosis with disorganized appearance. In the spine, involved vertebrae show an increased density, leading to the use of the term “ivory vertebrae.”
These three phases were initially described as unique entities using plain radiographs. However, with the advent of computed tomography (CT) and magnetic resonance imaging (MRI), it appears that these phases occur along a continuum and may co-exist at involved sites. In addition, this classification system is not easily applied to the spine.
Advanced Imaging
Dohan et al. reviewed the CT and MRI appearance of Paget’s and its differing appearance from other vertebral lesions.42 They describe the CT scan patterns falling into three categories: bone texture, bone marrow and vertebral shape.
Diffuse bone texture changes are described as occurring in three patterns. The first is the multicystic pattern. Here, soft tissue densities or cystic spaces in the body and neural arch replace cancellous bone. The second is a mesh pattern, characterized by an interweaving of scarce trabeculae. The final pattern is diffusely sclerotic. This is characterized by a homogenous formation of bone without an increase in the bone’s size. This leads to the characteristic finding of ivory vertebrae. When the sclerotic pattern extends into the posterior elements, this finding is strongly suggestive of PD. Focal bone texture changes may also be present including sclerotic foci, bone-within-bone appearance and horizontal course trabeculae. With the horizontal course trabeculae converging toward the pedicle and the sclerotic endplates, a classic “picture frame sign” can be seen on sagittal CT sections.
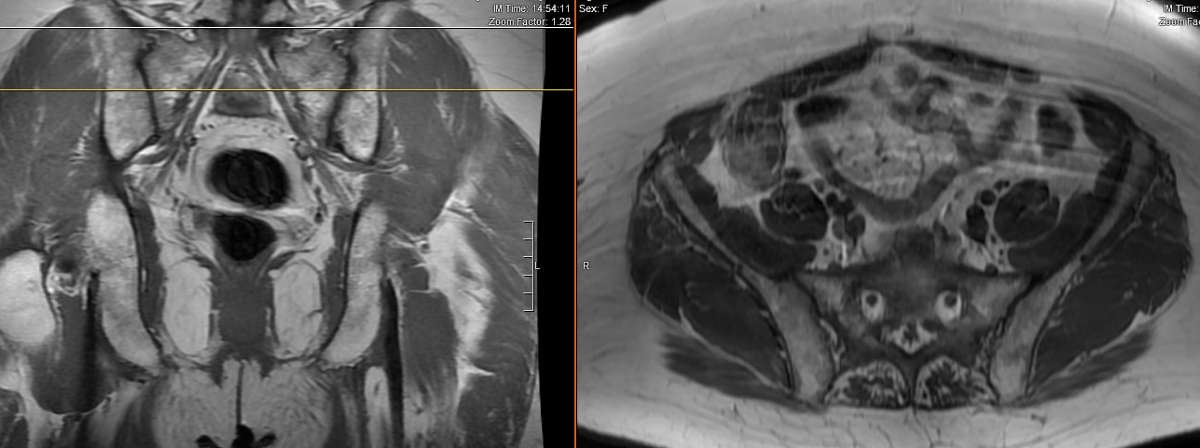
There can be some overlap of appearance of the vertebrae in Paget’s disease with other conditions such as hemangioma, metastatic disease or lymphoma. MRI can be indispensable in ruling out these other causes. In the intermediate stage of PD, there may be a high amount of bone marrow edema, with high signal intensity on T2-weighted images and low signal intensity with post-gadolinium enhancement on T1-weighted images. During the sclerotic phase, the bone marrow has become mainly fibrotic with significant replacement due to diffuse trabecular thickness, resulting in low signal intensity on both T1 and T2 images. During late stages of PD, following the completion of bone texture and shape changes, there is a repopulation of normal marrow. There is a uniform high signal intensity on T1- and T2-weighted images and low signal intensity on fat-saturated T2-weighted images (See Figs. 3-2A and B).

NON-OPERATIVE TREATMENT
Pharmacologic Therapies and Outcomes
The goal of pharmacologic treatment of Paget’s is to inhibit the osteoclastic bone resorption and resulting disorganized bone formation. Many different medications have been developed and implemented over the past several decades for treatment of Paget’s disease. Initial medications included calcitonin, mithramycin, gallium nitrate and ipriflavone.43 These, however, were largely abandoned following the successful development and widespread implementation of bisphosphonates. Nitrogen containing bisphosphonates work to inhibit osteoclast function via binding and blocking the enzyme farnesyl diphosphate synthase (FPPS) in the HMG-CoA reductase/mevalonate pathway. Current indications for initiating medical treatment with bisphosphonates include bone pain, hearing loss, high output heart failure, hypercalcemia secondary to immobilization and neurologic dysfunction. Although there is consensus on treatment of patients with symptomatic Paget’s disease, there appears to be far less consensus on the treatment of asymptomatic patients. With more frequent use of medical imaging, Paget’s disease may be incidentally diagnosed at earlier stages when the patient is asymptomatic. The frequently cited goal of treating these patients is to reduce the risk of developing complications with progression of Pagetic lesions, especially in worrisome regions such as the skull, spine and weight bearing long bones. These lesions are estimated to progress, on average, approximately one centimeter per year. The largest study evaluating this question of treating asymptomatic patients is the PRISM study by Langston et al.44 The authors evaluated 1,324 patients, on average, for three years, and 502 patients for an additional three years. Patients were randomized into either a symptomatic treatment group or intensive treatment group. The symptomatic group was treated with anti-inflammatory or analgesic medications on an as-needed basis. They were given bisphosphonates only if they did not respond to these first line treatments. The intensive treatment group was given multiple courses of bisphosphonates until serum alkaline phosphatase returned to normal levels. The authors found that there was a significantly lower alkaline phosphatase level within four months of initiating therapy in the intensive treatment group. Despite this improvement in serum markers, they reported no difference between the groups in quality of life, in overall bodily pain or in Pagetic bone pain. Hearing thresholds also did not change significantly. Fractures occurred in 7.0% and 7.4% in the intensive and symptomatic treatment groups, respectively. In their follow-up study extending follow-up three additional years, these results were maintained. In addition, a non-significant increase in the risk of fractures was noted with intensive therapy. They concluded that bisphosphonate therapy should focus on control of symptoms and not suppression of bone turnover.
There have been several studies comparing different bisphosphonates and treatment regimens.45-49 However, many of these trials compared biochemical markers rather than symptoms or clinical outcomes. In general, these studies did show that use of most nitrogen containing bisphosphonates, with a variety of different regimens, were able to achieve biochemical remission in 75-95% of patients in 6-12 months.
OPERATIVE CARE
Back pain is the most common presenting symptom in Paget’s disease involving the spine. However, other complications can occur including lytic weakening and destruction of the vertebrae, as well as stenosis from vertebral expansion. Chronic back pain in Paget’s can generally be attributed to the hypervascularity seen with the pathologic remodeling process. This is usually reported as a deep dull ache that is not activity dependent. However, acute worsening of pain can frequently be a presenting symptom of acute vertebral compression fracture. Compression fractures are most commonly seen in the lumbar spine.50 When imaging these fractures with MRI, the fracture line will be low signal on T1. Acute fractures will show a high signal on T2 and short T1-inverse recovery (STIR) sequences. Initially, medical treatment with bisphosphonates may be attempted if there are no neurologic deficits. However, with continued pain, surgical intervention may be considered. Vertebroplasty or kyphoplasty is the procedure of choice to stabilize the vertebrae in hopes of reducing mechanical pain. These procedures have been shown to be safe and effective treatments in a wide variety of similar pathologic processes. In addition, vertebroplasty with polymethyl methacrylate (PMMA) may assist in stabilizing the anterior and middle column, should the patient require a decompression via removal of the posterior elements. However, one consideration must be made for Paget’s disease of the spine; the lesions are hypervascular, which increases risk and has a high association with excessive blood loss and increased mortality. Pretreatment with bisphosphonates may assist in reducing this hypervascularity. However, in the acute setting, embolization may also help to reduce this risk.
One of the defining characteristics of Paget’s disease is abundant bone turnover with expansion of bony structures. This can cause significant dysfunction in the spine. Compression can occur with stenosis of the spinal cord, cauda equina and even exiting nerve roots. A combination of imaging with MRI and CT can ideally demonstrate the cause and site of this compression. Since Paget’s disease affects all the structures of the vertebral column, stenosis is commonly a result of posterior expansion of the vertebral body as well inward expansion of the neural arch and facets.
Surgery for Paget’s disease associated neural compression should be specifically tailored to site of stenosis. If there is central stenosis caused by the dorsal expansion of the vertebral body, a vertebrectomy with cage and fusion should be considered. If it is the posterior elements causing a compression of neural elements, then a posterior decompression would be the ideal treatment. Fusion should be considered in cases with instability. However, note that autograft from bones affected by polyostotic Paget’s disease can translocate the disease to grafted sites.51
PAGETIC OSTEOSARCOMA
Although a rare complication, osteosarcoma transformation in Paget’s disease is a dreaded complication of the disease. In his original paper, Paget did describe this malignant transformation in several of his published cases.1 Subsequent studies have shown malignant transformation to occur in < 1% of cases.52 Cases generally occur in patients with a history of longstanding disease, usually in the 7th or 8th decade of life. Generally, patients will present with worsening of previously well-controlled pain. Imaging will frequently show an expanding lytic lesion in a previously Pagetic bone. Diagnosis can be confirmed with biopsy of the lytic site. Histopathology from biopsy generally shows similar chaotic bone remodeling to Paget’s; however, there is a high level of cellular pleiomorphism and occasional malignant osteoid. Fortunately, although Paget’s disease is common in the spine, malignant transformation in the spine is exceptionally rare.
Generally, the treatment of Paget’s osteosarcoma is similar to other osteosarcomas with neoadjuvant/adjuvant chemotherapy and resection. However, since this disease is limited to mostly elderly patients who may not be able to tolerate chemotherapy, even with resection of the disease the prognosis is uniformly fatal.
CONCLUSION AND FUTURE DIRECTIONS
Paget’s disease of the spine is a progressive disease without a fully understood root cause. Although its prevalence is decreasing globally, there are still several studies investigating its potential genetic and environmental causes. One of the most important developments in the diseases’ history has been the use of bisphosphonate therapy for the treatment of symptomatic patients. Surgery can play an important adjuvant role as well. However, this is usually limited to decompression and stabilization, similar to other degenerative conditions in the spine. Current ongoing genetic studies may lead to a potential target for directed therapy that may eventually lead to eradication of this disease.
REFERENCES
- Paget J. Chronic inflammation of bones (Osteitis Deformans). Tr. Roy. Med-Chir. Soc., Glasgow. 1877;60:37-63.
- O’Driscoll JB, Anderson DC. Past Pets and Paget’s disease. Lancet. 1985;2(8461):919-921.
- Siris ES, Kelsey JL, Flaster E, Parker S. Paget’s disease of bone and previous pet ownership in the United States: dogs exonerated. Int J Epidemiol. 1990;19(2):455-458.
- Khan SA, Brennan P, Newman J, Gray RE, McCloskey EV, Kanis JA. Paget’s disease of bone and unvaccinated dogs. Bone. 1996;19(1):47-50.
- Mee AP, Dixon JA, Hoyland JA, Davies M, Selby PL, Mawer EB. Detection of Canine distemper virus in 100% of Paget’s disease samples by in situ-reverse transcriptase-polymerase chain reaction. Bone. 1998;23(2):171-175.
- Gordon MT, Mee AP, Anderson DC, Sharpe PT. Canine distemper virus sequenced from pagetic bone. Bone and Mineral. 1992;19(2):159-174.
- Selby PL, Davies M, Mee AP. Canine distemper virus induces human osteoclastogenesis through NF-KB and sequestosome 1/P62 activation. J Bone Miner Res. 2006;21(11):1750-1756.
- Lever JH. Paget’s disease of bone in Lancashire and arsenic pesticide in cotton mill wastewater: a speculative hypothesis. Bone. 2002;31(3):434-436.
- Rebel A, Malkani K, Basle J. Anomalies nuleaires des osteoclasts de la maladie oseeuse de Paget. Nouv Presse Med. 1974;3:1299-1301.
- Mills BG, Singer FR. Nuclear inclusion in Paget’s disease of bone. Science. 1976;194:201-202.
- Dickson GR, Shirodria PV, Kanis JA, Beneton MNC, Carr KE, Mollon RAB. Familial expansile osteolysis: A morphological, histomorphometric and serological study. Bone. 1991;12(5): 331-338.
- Beneton MN, Harris S, Kanis JA. Paramyxovirus-like inclusions in two cases of pycnodysostosis. Bone.1987;8(4):211-217.
- Mills BG, Yabe H, Singer FR. Osteoclasts in human osteopetrosis contain viral-nucleocapsid-like nuclear inclusions. J Bone Miner Res. 1988;3:101-106.
- Mills B, Singer FR, Weiner L, Suffin S, Stabile E, Holst P. Evidence for both respiratory syncytial virus and measles virus antigens in the osteoclasts of patients with Paget’s disease of bone. Clin Orthop Relat Res. 1984;183(3):303-311.
- Basle MF, Fournier JG, Rozenblatt S, Rebel A, Bouteille M. Measles virus RNA detected in Paget’s disease bone tissue by in situ hybridization. J Gen Virology. 1986;67(5):907-913.
- Friedrichs WE, Reddy SV, Bruder JM, et al. Sequence analysis of measles virus nucleocapsid transcripts in patients with Paget’s disease. J Bone Miner Res. 2002;17:145-151.
- Mee AP, Hoyland JA, Baird P, Bennett D, Sharpe PT. Canine bone marrow cell cultures infected with canine distemper virus: an in vitro model of Paget’s disease. Bone. 1995;17(4):S461-466.
- Mills BG, Frausto A, Singer FR, Ohsaki Y, Demulder A, Roodman GD. Multinucleated cells formed in vitro from Paget’s bone marrow express viral antigens. Bone. 1994;15(4): 443-448.
- Rivera-Toledo E, Gomez B. Respratory scyncytial virus persistence in macrophages alters the profile of cellular gene expression. Viruses. 2012;4(12):3270-3280.
- Ralston SH, Albagha OM. Genetics of Paget’s disease of bone. Curr Osteoporos Rep. 2014;12(3):263-271.
- Sofaer JA, Holloway SM, Emery AE. A family study of Paget’s disease of bone. J Epidemiol Community Health. 1983;37:226-231.
- Siris ES, Ottman R, Flaster E, Kelsey JL. Familial aggregation of Paget’s disease of bone. J Bone Miner Res. 1991;6(5):495-500.
- Hocking L, Slee F, Haslam SI, Cundy T, Nicholson G, van Hul W, Ralston SH. Familial Paget’s disease of bone: patterns of inheritance and frequency of linkage to chromosome 18q. Bone. 2000;26(6):577-580.
- Laurin N, Brown JP, Lemainque A, et al. Paget disease of bone: mapping of two loci at 5q35-qter and 5q31. Am J Hum Genet. 2001;69(3):528-543.
- Hocking LJ, Herbert CA, Nicholls RK, et al. Genomewide search in familial Paget disease of bone shows evidence of genetic heterogeneity with candidate loci on chromosomes 2q36, 10p13, and 5q35. Am J Hum Genet. 2001;69(5):1055-1061.
- Goode A, Long JE, Shaw B, et al. Paget disease of bone-associated UBA domain mutations of SQSTM1 exert distinct effects on protein structure and function. Biochim Biophys Acta. 2014;1842(7):992-1000.
- Wright T, Rea SL, Goode A, et al. The s349T mutation of SQSTM1 links Keap1/Nrf2 signaling to Paget’s disease of bone. Bone. 2013;52(2):699-706.
- Sundaram K, Shanmugarajan S, Rao DS, Reddy SV. Mutant p62P392L stimulation of osteoclast differentiation in Paget’s disease of bone. Endocrinology. 2011;152(11):4180-4189.
- Jin W, Chang M, Paul EM, et al. Deubiquinating enzyme CYLD negatively regulates RANK signaling and osteoclastogenesis in mice. J Clin Invest. 2008;118(5):1858-1866.
- Chamoux E, Couture J, Bisson M, Morissette J, Brown JP, Roux S. The p62 P392L mutation linked to Paget’s disease induces activation of human osteoclasts. Mol Endocrinol. 2009;23(10):1668-1680.
- Albagha OME, Wani S, Visconti MR, et al. Genome-wide association study identifies three new susceptibility loci for Paget’s disease of Bone. Nat Genet. 2011;43(7):685-689.
- Albagha OM, Visconti MR, Alonso N, et al. Genome-wide association study identifies variants at CSF1, OPTN, and TNFRSF11A as genetic risk factors for Paget’s disease of bone. Nat Gent. 2010;42:520-524.
- Albagha OM, Visconti MR, Alonso N, et al. Common susceptibility alleles and SQSTM1 mutations predict disease extent and severity in a multinational study of patients with Paget’s disease. J Bone Miner Res. 2013;28(11):2238-2246.
- National Institute of Health: U.S. National Library of Medicine. Clinical Trials.gov. Zoledronate in the Prevention of Paget’s Disease: Long Term Extension (ZiPP-LTE). https://clinicaltrials.gov/ct2/show/NCT03859895. Accessed on July 24, 2019.
- Rogers J, Jeffrey DR, Watt I. Paget’s disease in an archeological population. J Bone Miner Res. 2002;17(6):1127-1134.
- May S. Archeological skeletons support a northwestern European origin for Paget’s disease of bone. J Bone Miner Res. 2010; 25(8):1839-1841.
- Corral-Guddino L, Borao-Cengotita-Bengoa M, Del Pino-Montes J, Ralston S. Epidemiology of Paget’s disease of bone: a systematic review and meta-analysis of secular changes. Bone. 2013; 55(2);347-352.
- Cundy T. Is the prevalence of Paget’s disease of bone decreasing? J Bone Miner Res. 2006;21 Suppl 2:P9-13.
- Cooper C, Schafheutle K, Dennison E, Kellingray, P, Barker D. The epidemiology of Paget’s disease in Britain: is the prevalence decreasing? J Bone Miner Res. 1999; 14(2):192-197.
- Hadjipavlou A, Lander P. Paget disease of the spine. J Bone Joint Surg Am. 1991;73(9):1376-1381.
- Altman RD, Bloch DA, Hochberg MC, Murphy WA. Prevalence of pelvic Paget’s disease of bone in the United States. J Bone Miner Res. 2000;15(3):461-465.
- Dohan A, Parlier-Cuau C, Kaci R, Touraine S, Bousson V, Laredo JD. Vertebral involvement in Paget’s disease: morphological classification of CT and MR appearances. Joint Bone Spine. 2015;82(1):18-24.
- Hadjipavlou AG, Gaitanis IN, Kontakis GM. Paget’s disease of the bone and its management. J Bone Joint Surg Br. 2002;84(2):160-169.
- Langston AL, Campbell MK, Fraser WD, et al. Randomized trial of intensive bisphosphonate treatment versus symptomatic management in Paget’s disease of bone. J Bone Miner Res. 2010;25(1):20-31.
- Miller PD, Brown JP, Siris ES, Hoseyni MS, Axelrod DW, Bekker PJ. A randomized, double-blind comparison of risedronate and etidronate in the treatment of Paget’s disease of bone. Am J Med 1999;106(5):513-520.
- Siris E, Weinstein RS, Altman R, et al. Comparative study of alendronate versus etidronate for the treatment of Paget’s disease of bone. J Clin Endocrinol Metab. 1996;81(3):961-7
- Merlotti D, Gennari L, Martini G, Vallegi F, De Paola V, et al. Comparison of different intravenous bisphosphonate regimens for Paget’s disease of bone. J Bone Miner Res. 2007;22(10):1510-1517.
- Hosking D, Lyles K, Brown JP, et al. Long term control of bone turnover in Paget’s disease with Zoledronic acid and risedronate. J Bone Miner Res 2007;22(5):142-148.
- Reid IR, Hosking DJ. Bisphosphonates in Paget’s disease. Bone 2011;49(1):89-94.
- Boutin RD, Spitz DJ, Newman JS, Lenchik L, Steinbach LS. Complications in Paget disease at MR imaging. Radiology 1998;209(3):641-651.
- Hamadouche M, Mathieu M, De Pinieux G, Topouchian V, Courpied JP. Paget’s disease of bone can be transferred through autogenous bone grafting. Poster #1033. Orthopaedic Research Society Annual Meeting. New Orleans, LA:2003.
- Schajowicz F, Santini Araujo E, Berenstein M. Sarcoma complicating Paget’s disease of bone. A clinicopathological study of 62 cases. J Bone Joint Surg Br. 1983;65(3):299-307.