
Minna Kraatari, Sini Skarp, Minna Männikkö, Jaro Karppinen, Frances Williams
INTRODUCTION TO GENETICS
The genome is the complete set of genetic information stored as deoxyribonucleic acids (DNA) in the nucleus of cells. The nucleic acid sequence of the genome determines the recipe for life: the instructions for each protein and ribonucleic acid (RNA) are stored there as genes. Both forms of nucleic acid – RNA and DNA – have a backbone consisting of sugars and phosphates to which the bases are attached. In DNA the sugar in the sugar-phosphate-backbone is deoxyribose and in RNA ribose. The four bases in DNA are thymine (T), adenine (A), guanine (G) and cytosine (C). The C and G, as well as A and T bases, are always paired together forming base pairs (bp) when DNA forms its typical double helix structure (Fig. 1-1). DNA is more stable of the two nucleic acids and thus is well suited to its role in long-term storage of genetic data. The forms of RNA, on the other hand, have a more short-term role in the cell.

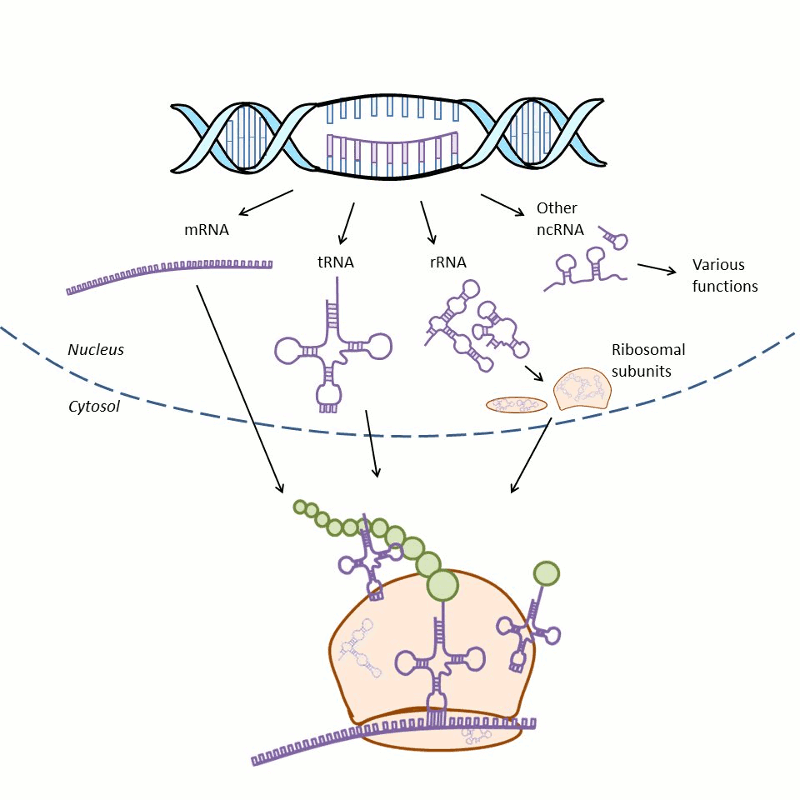
RNAs perform many functions during protein synthesis and in the regulation of gene expression (Fig. 1-2). During protein synthesis, the genetic information of a gene is first copied into messenger RNA (mRNA) in a process called transcription and then into protein via a process termed translation. During translation, the transfer RNA (tRNA) provides amino acids to the ribosome that synthesizes the primary structure of proteins. Ribosomes consist of proteins and ribosomal RNA (rRNA). There is also a great variety of noncoding RNAs (ncRNA) that regulate gene expression.
Genes are units in the genome that encode RNA as either messenger RNA (mRNA), transfer RNAs (tRNA), ribosomal RNAs (rRNA) or noncoding RNAs (ncRNA). A protein coding gene typically consists of a promoter region, exons, introns and variety of regulation sites called enhancers and silencers located further away in the genome. Exons encode the protein sequence. Introns are regions between the exons that are originally included into the mRNA but are later removed in a process called splicing. Promoter regions are near the transcription start site and are bound by transcription factors and RNA synthesizing enzymes (RNA polymerase) at the beginning of transcription. Enhancers and silencers can be located a long way – several hundred thousand base pairs upstream or downstream – from the transcription start site. Binding of transcription factors to these sites may increase (enhancers) or decrease (silencers) gene expression.
The place in the genome where a gene is found is known as the locus. Comparison of genomes between people, even in the absence of disease, reveals variations in the base pair sequence. Such differences may be deletions, insertions, inversions or substitutions of one or several base pairs. A substitution of one base pair with another is called a single nucleotide polymorphism (SNP). When such a variant falls within a coding region, the gene is said to possess several ‘alleles.’ While at the population level a given gene may have several alleles, a person has two alleles, one inherited from the mother and the other from the father. When an individual has similar alleles from both parents, they are considered homozygous, and when the two alleles are different, they are heterozygous for the allele.
The human genome consists of more than three billion base pairs that are packed tightly by wrapping the DNA molecules around histone proteins and stored as 46 chromosomes. Around 20,000 protein-coding genes are estimated to be found in human genome making approximately 1-2% of the genome. The rest of the DNA consists of regulatory sequences (~20%), so-called repetitive DNA (~50%), introns (~5%) and other non-coding regions.
The variation of the human genome is approximately 0.1% between individuals; nevertheless, each person carries millions of variations in their genome. These variations form the basis for much of the difference between individuals from the color of eyes to the predisposition to disease. In complex traits influenced by both genetic and environmental factors, such as height or intervertebral disc degeneration, the outcome is affected by the genetic variation as well as other factors including the environment and lifestyle. The contribution of the genetic component in each trait can be measured at the population level, and this proportion of variation attributable to genetic factors is referred to as the heritability of a trait.
In complex disease, the genetic component typically comprises many different variants in different genes that all contribute a small amount to the overall phenotype. The variation may be divided into common and rare variation based on the minor allele frequency (MAF). A typical definition is that variants having MAF over 5% are considered common variants and variants having MAF less than 1% are rare variants. Variants with MAF between 1% and 5% are considered intermediate frequency variants.
METHODS FOR STUDYING GENETIC FACTORS
Different genes and different variants with different frequencies and effect sizes affect the predisposition to complex diseases together. A common variation is likely to cause subtle effects shared by populations whereas rare variants can have a larger effect. The smaller the effect of the variant the larger the sample from the population is needed to detect it. Family-based studies focusing on extreme phenotypes or rare forms of common diseases can be used to identify rare variants. Genome-wide association studies (GWAS) with thousands, tens of thousands or even hundreds of thousands of subjects look for common predisposing variants shared by populations.
The candidate gene approach studies genes that can be considered candidates based on their function and previous knowledge of the biology of disease. Typically, the predetermined genetic regions are studied using case-control setting to identify alleles which are significantly more or less common in the cases than in controls. The genetic regions can be a gene(s), parts of gene or candidate variants.
Linkage analysis has been used to study complex diseases before the advent of GWAS. Linkage analysis is most powerful when used to study rare variants and when the inheritance pattern is known. The ideal study population is a large extended family. This is not usually the case with complex diseases. In a linkage study multiple variants, usually SNPs or short repeats (microsatellites) spanning the genome, are used as genetic markers. These markers are compared between the affected and unaffected individuals. The loci are in linkage when they are inherited together more often than would be expected in the case of independent inheritance.
The arrival of GWAS, made possible by the International HapMap Project, was one of the most important technical achievements in the genetics of complex diseases. GWAS typically used tagged variants to study the entire genome in large samples of unrelated individuals. Hundreds of thousands of common variants, typically SNPs, are compared in either a case and control study design, or quantitative trait, to identify genomic regions that harbor variants with statistically different frequencies between the groups. Huge international consortia were established, and in a few years, GWAS had identified thousands of associations between complex disorders and genetic markers. The associations typically do not point to the disease-causing or ‘functional’ SNP but rather to the region harboring such which are ‘tagged’ by the associating SNP(s). Further studies are performed to identify the functional link between the variant and the disease. Despite the successes in the decade following the first GWASs in 2005, such as the discovery of the FTO gene in obesity1 only relatively small amount of the heritability in common disease has been accounted for by common variants.
The other big technical step in genetics was the maximum parallel sequencing also known as next generation sequencing (NGS) that lowered the price of sequencing. The different approaches using NGS have been targeted re-sequencing, whole exome sequencing and whole genome sequencing. The targeted re-sequencing means capturing multiple regions in the genome that are previously associated with the phenotype. Whole exome sequencing means sequencing of all the protein-coding areas (i.e., exons) in the genome. NGS methods have been successfully used to study Mendelian traits2 and complex diseases,3 as well as cancer.4
HERITABILITY OF INTERVERTEBRAL DISC DEGENERATION
Intervertebral disc degeneration (DD) is considered a condition that is in large part genetically determined with environmental factors also playing a role.5 It was previously thought that age, gender, occupational exposures, mechanical influences, smoking and lifestyle factors mostly contributed to DD and disc herniation.6,7 Since then, the heritability of DD has been studied widely, and twin and familial studies suggest that genetic factors contribute up to 75% of the variance in DD in populations, whereas environmental factors, such as occupational vibration exposure and smoking, have only modest effect.8-10
Heritability of DD varies between studies according to the specific feature on spine MR studied. Heritability of disc bulge is estimated to be 65%, and heritability of disc space narrowing is estimated being 79%.5 Modic changes (MC), pathological bone marrow signal changes adjacent to vertebral endplate visible only on MRI, are a distinct phenotype of DD. The heritability of MC is estimated to be 16-34%.11 Overall the heritability estimates show that the genetic component is significant enough to be studied.
GENES ASSOCIATED WITH DISC DEGENERATION
Genes associated with DD may be grouped into the following categories based on their function in the disc; genes affecting the structure of the intervertebral disc (IVD), genes responsible for producing enzymes of the disc matrix, inflammatory genes and genes with different functions (Table 1-1). Identification of the genes affecting the development of DD has provided a key insight into the molecular mechanism of the disease, although many of the previously identified variants are likely to be false positive as is clearly demonstrated for bone mineral density.12 Meta-analysis has found moderate support for only a handful of genes (see Table 1-1).
| Gene | Gene Name | Category |
|---|---|---|
| Type I collagen | COL1A1 | Structural |
| Type IX collagen | COL9A1, COL9A2, COL9A3 | |
| Type XI collagen | COL11A1, COL11A2 | |
| Aggrecan | ACAN | |
| Asporin | ASPN | |
| Cartilage intermediate layer protein | CILP | |
| Thrombospondins | THBS | |
| Matrix metalloproteases 1 | MMP1 | Catabolic |
| Matrix metalloproteases 2 | MMP2 | |
| Matrix metalloproteases 3 | MMP3 | |
| Parkinson protein 2 | PARK2 | |
| Proteosome subunit β type 9 (P) | PSMB9 | |
| Carbohydrate sulfotransferase 3 | CHST3 | |
| Tissue inhibitors of metalloproteinases | TIMP | Anti-catabolic |
| Growth differentiation factor 5 | GDF5 | |
| Interleukin-1 | IL1 | Inflammatory |
| Interleukin-6 | IL6 | |
| Cyclooxygenase-2 | COX2 | |
| Vitamin D receptor | VDR | Other |
| Sickle tail | SKT |
Structural Genes
The extracellular matrix (ECM) is composed of three major classes of molecules; structural proteins such as collagens, fibrillins and elastin; specialized proteins such as fibronectin, laminins and integrins; and proteoglycans. Collagens are the most abundant proteins in the human body found, for example, in tendons, ligaments, skin, cartilage, bones and IVD. There are 44 collagen genes that scatter throughout the human genome combining over 28 different types of collagen fibrils. Proteoglycans are composed of a core protein and glycosaminoglycan chains extending perpendicularly from the core protein in a bottle brush-like structure. There are a huge number of different proteoglycans varying by core protein and the number and type of glycosaminoglycan chain. They interact with the other proteins in the ECM and keep the level of fluidity high providing resistance to compressive loads.
Type I collagen is an important protein in the ligaments, skin and bone encoded by two genes; COL1A1 and COL1A2. It is a predominant component of outer annulus fibrosus providing tensile strength to resist the tensile, shear and torsional loads.13 A promoter region G to T SNP called Sp1 (rs1800012) of COL1A1 has been reported to increase the risk of many bone related conditions such as osteoporosis and increased risk of fractures.14 In a rare bone fragility condition, osteogenesis imperfecta, variants in these two genes account for the majority of cases. Although the mechanism by which polymorphisms influence the development of DD is not fully understood, the TT genotype of COL1A1 was found to be a risk factor for DD.15 It is suggested that T allele leads to increased collagen α 1(I) chain and collagen α 1(I)/α 2(II) ratio. The increase of collagen α 1(I) chain has been associated with impaired bone strength and thus is thought to impair the disc structure.16
Type IX collagen is found in annulus fibrosus, nucleus pulposus and the endplate of the disc and is encoded by the COL9A1, COL9A2 and COL9A3 genes. The function of type IX collagen is not clearly understood, but it provides mechanical support to the disc.17 A rare allele of COL9A2, Trp2 (Gln326Trp), was first found to be associated with DD in Finnish sciatica patients. The finding has been replicated;18,19 however, controversial results have also been obtained.20 Another polymorphism, Trp3 (Arg103Trp) of the COL9A3 gene, has been found to be associated with DD later studies confirmed.21,22 The Trp2 and Trp3 associations are among the most strongly correlated relationships with DD, and the results have been replicated in more than two different ethnic populations. The increased risk may be explained by the addition of tryptophan to the collagen chain, an amino acid rarely found in collagenous domains which may impair collagens’ mechanical properties and thus weaken the disc resistance to compressive loads.20
Type XI collagen is minor collagen in the IVD but is essential in the interaction between proteoglycans and collagens.21 It is encoded by the COL11A1, COL11A2 and COL11A3 genes. There are several collagen 11 SNPs that have been associated with DD.19,22 However, none of the results have so far been replicated in different populations.19 Further, the functional roles of these SNPs are still unknown. It is suggested that they might produce unstable transcripts of the disease-associated allele which would cause decreased functional collagen and lead to degradation.19 Mutations in the COL2A1, COL9A1, COL11A1 or COL11A2 genes have also been associated with autosomal dominant Stickler syndrome and an autosomal recessive form is caused by mutations in the COL9A1 gene. Stickler syndrome is an inherited connective tissue disorder that affects the cartilage, articular tissues and eyes.23,24
Aggrecan is a large proteoglycan responsible for the ability of the tissue to resist compressive loads.25 Aggrecan has a large number of chondroitin sulfate chains present in its core protein located in two adjacent regions; CS1 and CS2. The aggrecan gene (ACAN) carries variable number (from 13 to 33) of tandem repeat polymorphisms in the part encoding the CS1 domain.19 This is thought to result in variation in the degree of chondroitin sulfate substitution of aggrecan. Studies suggest that individuals with fewer repeats have less chondroitin sulfate and thus may be susceptible to premature degeneration.25 The association between this polymorphism and DD was first studied in Japanese women with A18 (18 repeats) and A21 (21 repeats) alleles over presented with multilevel and severe degeneration.26 Later, a Finnish study discovered that carrying two copies of A26 (26 repeats) allele associated with low back pain (LBP) and decreased intervertebral disc signal intensity on MRI.27 Also, C to T SNP (rs1042631) was found to be associated with signal intensity and disc bulge was found while G to T SNP (rs1516797) was found to associate with disc height narrowing.22
Asporin is a member of the family of small leucine-rich proteoglycans. It is associated with ECM in cartilage and meniscus providing functional support and regulating signaling molecules.28 Asporin usually contains 13 aspartic acid repeats at the amino-terminal, but a polymorphic allele at this site can contain 9-20 aspartic acid repeats.28 The polymorphic allele with 14 aspartic acid repeats has been shown to be associated with osteoarthritis and DD.19,29
Cartilage intermediate layer protein (CILP) is widely expressed in the IVD and acts as a negative regulator of transforming growth factor beta 1 (TGF-β), potentially down regulating aggrecan and type II collagen.30 C to T SNP resulting in an amino acid substitution (Ille395Thr) in exon 8 has been shown to associate with DD.30 The C allele was found to be the risk allele resulting in increased binding and inhibition of TGF-β.30 However, this association has not been discovered in any other population samples suggesting that either these results are false positives or that there may be unique genetic risk factors in different populations.
Thrombospondins are extracellular glycoproteins that bind to collagen and take part in cell-to-cell and cell-to-matrix communications. They have diverse functions related to the extracellular matrix and aid regulating the levels of MMP2 and MMP9.31 Polymorphisms in the THBS2 gene have been found to be associated with DD.19
Catabolic Genes
Matrix metalloproteases (MMP) are key proteins involved in the breakdown of the disc matrix which is one of the important pathological processes in DD. They are also involved in various physiological and pathological processes including angiogenesis, tissue repair, arthritis and metastasis. At the moment there are 18 known members of the MMP family that are involved in the breakdown of the different matrix components.
Polymorphisms in the MMP1 and MMP2 genes have been found associated with DD. An insertion of guanine into the promoter region of MMP1 (rs1799750) was found more frequently from individuals without DD.32 The CC genotype of C to T SNP (rs243865) in MMP2 has been discovered to increase the risk of DD nearly 3-fold compared to the TT and CT genotypes.33
MMP3 is a proteoglycan-degrading enzyme induced by mechanical loading or inflammation. A promoter region polymorphism of the MMP3 gene produces alleles with five or six repeats of adenine (5A and 6A, respectively) with the 5A allele having twice the promoter activity as the 6A allele suggesting that the 5A phenotype might be associated with higher protein expression and thus higher IVD degeneration.34 Indeed, the 5A/6A polymorphism was linked to DD in a Japanese sample and later the results were replicated in a UK sample.35 However, no association with MMP alleles and DD was found in the Finnish Twin Spine Study.22 Also, A to G SNP (rs17576) of MMP9 was found to significantly associate with lumbar disc degeneration.36
The PARK2 gene encodes a protein called parkin and was found to be associated in the first GWAS meta-analysis of DD. Parkin is a component of the multiprotein E3 ubiquitin ligase complex which tags damaged proteins for proteosomal degradation. Previously mutations in the PARK2 gene were associated with juvenile Parkinson’s disease, Alzheimer’s disease, and several solid tumors. A to G SNP (rs926849) in PARK2 was found to be associated with DD in a GWAS meta-analysis of 4500 Northern Europeans.37 Also, two SNPs (rs2187689 and rs7767277) in proteasome subunit β type 9 (P) (PSMB9) gene were found to be associated with DD in the same study.37 PSMB9 is a proteasome that degrades unneeded or damaged proteins.19 This finding remains to be replicated in a non-Northern European population.
The carbohydrate sulfotransferase 3 (CHST3) gene encodes an enzyme responsible for catalyzing the sulfation of chondroitin. Rare mutations have been found in patients with recessive skeletal abnormalities.38 In a large linkage and association study A to G SNP (rs1245582) and A to C SNP (rs4148941) lying within a potential microRNA-513a-5p binding site, were found to be associated with DD in a large Asian sample using a combination of association and linkage methods.39
Anti-catabolic Genes
Tissue inhibitors of metalloproteinases (TIMP) is a group of protease inhibitors. The association of TIMP and DD has been studied in only one study so far. In this study, the researchers found an association between C to T SNP and a change in osteophyte grade.35
The growth differentiation factor 5 (GDF5) gene encodes a secreted ligand of the TGF-β superfamily of proteins and regulates the development of numerous tissue types including cartilage and joints. Mutations of GDF5 have also been associated with osteoarthritis. C to T SNP (rs143383) was found to be associated with combined phenotype of disc space narrowing and osteophytes among Northern European women.40
Inflammatory Genes
Interleukins are cytokines that response to stress and cause a range of systematic responses. Polymorphisms in a number of interleukins have been associated with osteoarthritis, rheumatoid arthritis, LBP, DD and sciatica.19 Interleukin-1 (IL-1) is a cytokine that is produced as a response to inflammation, injury or antigen challenge. There are three members of the IL-1 family: IL-1α, IL-1β and IL-1 receptor antagonist (IL-1RN). The first two are strong inducers of inflammation whereas IL-1RN is a suppressor of IL-1. Increased activity of IL-1 is related to tissue destruction and thus degenerate discs produce increased amounts of IL-1. IL-1 also increases MMP3, MMP9 and MMP13 gene expression and decreases the expression of normal ECM molecule genes.41 An SNP in IL-1α (rs1800587) has been found to be associated with DD: the TT genotype increased the risk of DD and disc bulge.42 In another study, C to T SNP in IL-1α was found to be associated with Modic change (MC) with T allele increasing the risk of MC by 2.5-fold.43 Interleukin-6 (IL-6) is a pro-inflammatory cytokine that can also act as an anti-inflammatory cytokine. Many IL-6 polymorphisms have been associated with DD.19
The cyclooxygenase-2 (COX2) gene encodes an enzyme participating in the synthesis of prostaglandins, prostacyclin, and thromboxane. An SNP (rs5277) in the COX2 gene was found to be associated with Kellgren-Lawrence degeneration grade on plain radiographs.35
Other Genes
The vitamin D receptor (VDR) mediates the function of vitamin D and is involved in bone mineralization and remodeling.44 VDR was the first candidate gene found to be associated with DD.45 Since then, the relationship between two VDR polymorphism (TaqI and FokI) and DD has been studied extensively and the association has been validated in large populations of different ethnic backgrounds including Chinese, Japanese, Finnish and English.46 The first polymorphism, TaqI is located in the noncoding region of exon 9, and the t-allele has been shown as a risk factor for DD.47 The second polymorphism, FokI, is located in exon 2 of VDR. The FokI polymorphism eliminates the first ATG translation initiation codon and allows the second translation codon to be translated. Therefore, there are two possible protein forms.48 The minor allele f was associated with decreased disc signal among middle-aged Finnish male twins.45 VDR polymorphisms did not feature in the systematic review by Eskola et al.29 This may be due to the risk of false positive findings due to the small sample sizes used in candidate genes studies.
The Sickle tail (SKT) protein is expressed in the human IVD. The G allele of SNP (rs16924573) of the SKT gene was found to be strongly associated with lumbar disc herniation in Japanese subjects, and the finding was replicated among Finnish subjects.49
CONCLUSION AND FUTURE DIRECTIONS
The theory of the origin of DD has evolved from an occupational and lifestyle theory to a highly heritable condition with multiple- genetic factors as well as environmental factors contributing. A number of genetic factors associated with DD have been discovered in both humans and animals. However, the exact mechanisms of the genetic defects are not yet fully understood. In a systematic review by Eskola et al., none of the genetic variants reached a level of strong association. A moderate level of evidence was found for variants in the asporin (ASPN), collagen type XI alpha 1 (COL11A1), growth differentiation factor 5 (GDF5), sickle tail (SKT), thrombospondin 2 (THBS2) and matrix metalloproteinase 9 (MMP9) genes.29 Also, other genes have been suggested to relate to the development of lumbar disc degeneration. However, there is only a weak level of evidence for these genes due to variation between study designs, populations, sampling methods and phenotype definition. Collaboration studies with large cohorts and well-defined phenotype are needed to understand the underlying genetics in DD.
REFERENCES
- Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316(5826):889-894.
- Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42(1):30-35.
- Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13(3):255-262.
- Welch JS, Westervelt P, Ding L, et al. Use of whole-genome sequencing to diagnose a cryptic fusion oncogene. JAMA. 2011;305(15):1577-1584.
- Battie MC, Videman T, Kaprio J, et al. The twin spine study: contributions to a changing view of disc degeneration. Spine J. 2009;9(1):47-59.
- Videman T, Battie MC. The influence of occupation on lumbar degeneration. Spine (Phila Pa 1976). 1999;24(11):1164-1168.
- Battie MC, Videman T. Lumbar disc degeneration: epidemiology and genetics. J Bone Joint Surg Am. 2006;88 Suppl 2:3-9.
- Sambrook PN, MacGregor AJ, Spector TD. Genetic influences on cervical and lumbar disc degeneration: a magnetic resonance imaging study in twins. Arthritis Rheum. 1999;42(2):366-372.
- Battie MC, Videman T, Gibbons LE, Fisher LD, Manninen H, Gill K. 1995 Volvo Award in clinical sciences. Determinants of lumbar disc degeneration. A study relating lifetime exposures and magnetic resonance imaging findings in identical twins. Spine (Phila Pa 1976). 1995;20(24):2601-2612.
- Bijkerk C, Houwing-Duistermaat JJ, Valkenburg HA, et al. Heritabilities of radiologic osteoarthritis in peripheral joints and of disc degeneration of the spine. Arthritis Rheum. 1999;42(8):1729-1735.
- Määttä JH, Kraatari M, Wolber L, et al. Vertebral endplate change as a feature of intervertebral disc degeneration: a heritability study. Eur Spine J. 2014;23(9):1856-1862.
- Richards JB, Kavvoura FK, Rivadeneira F et al. Collaborative meta-analysis: associations of 150 candidate genes with osteoporosis and osteoporotic fracture. Ann Intern Med. 2009;151(8):528-537.
- Antoniou J, Steffen T, Nelson F, et al. The human lumbar intervertebral disc: evidence for changes in the biosynthesis and denaturation of the extracellular matrix with growth, maturation, ageing, and degeneration. J Clin Invest. 1996;98(4):996-1003.
- Keen RW, Woodford-Richens KL, Grant SF, Ralston SH, Lanchbury JS, Spector TD. Association of polymorphism at the type I collagen (COL1A1) locus with reduced bone mineral density, increased fracture risk, and increased collagen turnover. Arthritis Rheum. 1999;42(2):285-290.
- Pluijm SM, van Essen HW, Bravenboer N, et al. Collagen type I alpha1 Sp1 polymorphism, osteoporosis, and intervertebral disc degeneration in older men and women. Ann Rheum Dis. 2004;63(1):71-77.
- Käpyla J, Jaalinoja J, Tulla M, et al. The fibril-associated collagen IX provides a novel mechanism for cell adhesion to cartilaginous matrix. J Biol Chem. 2004;279(49):51677-51687.
- Jim JJ, Noponen-Hietala N, Cheung KM, et al. The TRP2 allele of COL9A2 is an age-dependent risk factor for the development and severity of intervertebral disc degeneration. Spine (Phila Pa 1976). 2005;30(24):2735-2742.
- Annunen S, Paassilta P, Lohiniva J, et al. An allele of COL9A2 associated with intervertebral disc disease. Science. 1999;285(5426):409-412.
- Mayer JE, Iatridis JC, Chan D, Qureshi SA, Gottesman O, Hecht AC. Genetic polymorphisms associated with intervertebral disc degeneration. Spine J. 2013;13(3):299-317.
- Wrocklage C, Wassmann H, Paulus W. COL9A2 allelotypes in intervertebral disc disease. Biochem Biophys Res Commun. 2000;279(2):398-400.
- Maeda S, Ishidou Y, Koga H, et al. Functional impact of human collagen alpha2(XI) gene polymorphism in pathogenesis of ossification of the posterior longitudinal ligament of the spine. J Bone Miner Res. 2001;16(5):948-957.
- Videman T, Saarela J, Kaprio J, et al. Associations of 25 structural, degradative, and inflammatory candidate genes with lumbar disc desiccation, bulging, and height narrowing. Arthritis Rheum. 2009;60(2):470-481.
- Richards AJ, McNinch A, Martin H, et al. Stickler syndrome and the vitreous phenotype: mutations in COL2A1 and COL11A1. Hum Mutat. 2010;31(6):E1461-1471.
- Van Camp G, Snoeckx RL, Hilgert N, et al. A new autosomal recessive form of Stickler syndrome is caused by a mutation in the COL9A1 gene. Am J Hum Genet. 2006;79(3):449-457.
- Cs-Szabo G, Ragasa-San Juan D, Turumella V, Masuda K, Thonar EJ, An HS. Changes in mRNA and protein levels of proteoglycans of the anulus fibrosus and nucleus pulposus during intervertebral disc degeneration. Spine (Phila Pa 1976). 2002;27(20):2212-2219.
- Kawaguchi Y, Osada R, Kanamori M, et al. Association between an aggrecan gene polymorphism and lumbar disc degeneration. Spine (Phila Pa 1976). 1999;24(23):2456-2460.
- Solovieva S, Noponen N, Männikkö M, et al. Association between the aggrecan gene variable number of tandem repeats polymorphism and intervertebral disc degeneration. Spine (Phila Pa 1976). 2007;32(16):1700-1705.
- Lorenzo P, Aspberg A, Onnerfjord P, Bayliss MT, Neame PJ, Heinegard D. Identification and characterization of asporin. A novel member of the leucine-rich repeat protein family closely related to decorin and biglycan. J Biol Chem. 2001;276(15):12201-12211.
- Eskola PJ, Lemmela S, Kjaer P, et al. Genetic association studies in lumbar disc degeneration: a systematic review. PLoS One. 2012;7(11):e49995.
- Seki S, Kawaguchi Y, Chiba K, et al. A functional SNP in CILP, encoding cartilage intermediate layer protein, is associated with susceptibility to lumbar disc disease. Nat Genet. 2005;37(6):607-612.
- Bein K, Simons M. Thrombospondin type 1 repeats interact with matrix metalloproteinase 2. Regulation of metalloproteinase activity. J Biol Chem. 2000;275(41):32167-32173.
- Song YQ, Ho DW, Karppinen J, et al. Association between promoter -1607 polymorphism of MMP1 and lumbar disc disease in Southern Chinese. BMC Med Genet. 2008;9:38.
- Dong DM, Yao M, Liu B, Sun CY, Jiang YQ, Wang YS. Association between the -1306C/T polymorphism of matrix metalloproteinase-2 gene and lumbar disc disease in Chinese young adults. Eur Spine J. 2007;16(11):1958-1961.
- Kepler CK, Ponnappan RK, Tannoury CA, Risbud MV, Anderson DG. The molecular basis of intervertebral disc degeneration. Spine J. 2013;13(3):318-330.
- Valdes AM, Hassett G, Hart DJ, Spector TD. Radiographic progression of lumbar spine disc degeneration is influenced by variation at inflammatory genes: a candidate SNP association study in the Chingford cohort. Spine (Phila Pa 1976). 2005;30(21):2445-2451.
- Hirose Y, Chiba K, Karasugi T, et al. A functional polymorphism in THBS2 that affects alternative splicing and MMP binding is associated with lumbar-disc herniation. Am J Hum Genet. 2008;82(5):1122-1129.
- Williams FM, Bansal AT, van Meurs JB, et al. Novel genetic variants associated with lumbar disc degeneration in Northern Europeans: a meta-analysis of 4600 subjects. Ann Rheum Dis. 2013;72(7):1141-1148.
- Hermanns P, Unger S, Rossi A, et al. Congenital joint dislocations caused by carbohydrate sulfotransferase 3 deficiency in recessive Larsen syndrome and humero-spinal dysostosis. Am J Hum Genet. 2008;82(6):1368-1374.
- Song YQ, Karasugi T, Cheung KM, et al. Lumbar disc degeneration is linked to a carbohydrate sulfotransferase 3 variant. J Clin Invest. 2013;123(11):4909-4917.
- Williams FM, Popham M, Hart DJ, et al. GDF5 single-nucleotide polymorphism rs143383 is associated with lumbar disc degeneration in Northern European women. Arthritis Rheum. 2011;63(3):708-712.
- Millward-Sadler SJ, Costello PW, Freemont AJ, Hoyland JA. Regulation of catabolic gene expression in normal and degenerate human intervertebral disc cells: implications for the pathogenesis of intervertebral disc degeneration. Arthritis Res Ther. 2009;11(3):R65.
- Solovieva S, Kouhia S, Leino-Arjas P, et al. Interleukin 1 polymorphisms and intervertebral disc degeneration. Epidemiology. 2004;15(5):626-633.
- Karppinen J, Solovieva S, Luoma K, Raininko R, Leino-Arjas P, Riihimäki H. Modic changes and interleukin 1 gene locus polymorphisms in occupational cohort of middle-aged men. Eur Spine J. 2009;18(12):1963-1970.
- Haussler MR, Whitfield GK, Haussler CA, et al. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res. 1998;13(3):325-349.
- Videman T, Leppävuori J, Kaprio J, et al. Intragenic polymorphisms of the vitamin D receptor gene associated with intervertebral disc degeneration. Spine (Phila Pa 1976). 1998;23(23):2477-2485.
- Feng Y, Egan B, Wang J. Genetic factors in intervertebral disc degeneration. Genes Dis. 2016;3(3):178-185.
- Cheung KM, Chan D, Karppinen J, et al. Association of the Taq I allele in vitamin D receptor with degenerative disc disease and disc bulge in a Chinese population. Spine (Phila Pa 1976). 2006;31(10):1143-1148.
- Whitfield GK, Remus LS, Jurutka PW, et al. Functionally relevant polymorphisms in the human nuclear vitamin D receptor gene. Mol Cell Endocrinol. 2001;177(1-2):145-159.
- Karasugi T, Semba K, Hirose Y, et al. Association of the tag SNPs in the human SKT gene (KIAA1217) with lumbar disc herniation. J Bone Miner Res. 2009;24(9):1537-1543.